Ataxia es un término griego que significa “desorden”. Nos referimos a la ataxia como un signo clínico caracterizado por la descoordinación del movimiento: falta de estabilidad en la marcha; torpeza o debilidad en las extremidades superiores, inferiores, cuerpo o movimientos oculares, etc. como consecuencia de una afectación del Sistema Nervioso Central (SNC).
En líneas generales, la ataxia suele ser secundaria a una afectación del cerebelo o de sus vías nerviosas eferentes o aferentes, aunque otras estructuras cerebrales podrían ocasionar esta sintomatología. En este artículo repasaremos las características de este fenómeno.
Síntomas de la ataxia
Aunque las características principales de la ataxia son la descoordinación de las extremidades y de los movimientos sacádicos de los ojos, se pueden presentar otros tipos de síntomas. Todos los síntomas de la ataxia, sin embargo, tienen que ver con la capacidad para mover partes del cuerpo. Estas señales de que la ataxia está afectando a las funciones normales del cuerpo se describen a continuación.
-
Problemas del habla.
-
Dificultades en la percepción visuoespacial debido a la descoordinación oculomotora.
-
Apraxia visuoconstructiva como consecuencia de la descoordinación.
-
Disfagia –problemas para tragar–.
-
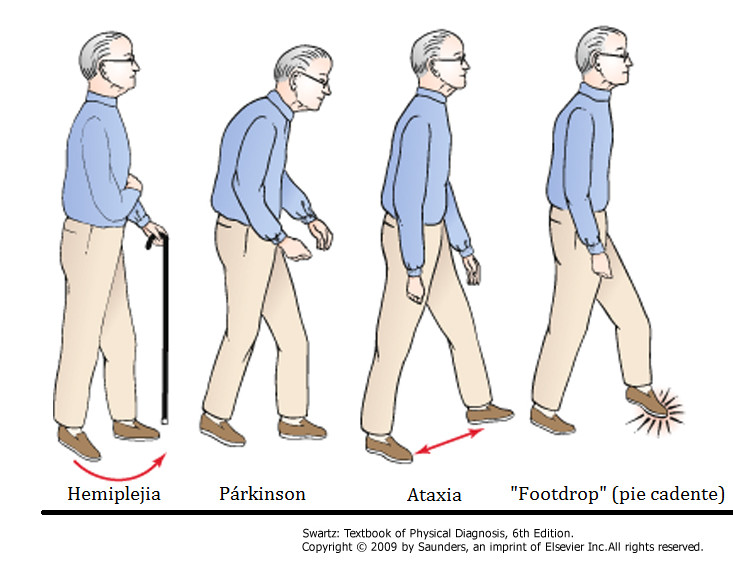
Dificultades en la marcha, con tendencia a abrir las piernas.
-
Pérdida total de la capacidad de andar.
Como hemos dicho, en la clínica, la ataxia suele presentarse como un signo que puede manifestarse en diferentes patologías adquiridas –es decir: infartos cerebrales, tumores, traumatismos cráneo-encefálicos, etc.– aunque también puede presentarse como una enfermedad aislada en sus formas hereditarias.
Clasificaciones (tipos de ataxias)
Podríamos clasificar la ataxia siguiendo diferentes criterios, aunque en esta revisión explicaremos los principales tipos de ataxia en función de si la patología ha sido adquirida o es hereditaria. Otro posible modo de clasificación sería en función de las regiones del Sistema Nervioso Central que presentan lesiones o anomalías susceptibles de producir ataxia.
1. Ataxias adquiridas
Que una ataxia sea adquirida implica que ésta se produce como consecuencia de una patología principal que sufre el paciente. De este modo los infartos cerebrales, la anoxia cerebral –falta de oxígeno en el cerebro–, tumores cerebrales, traumatismos, enfermedad desmielinizante –esclerosis múltiple–, son causas comunes de ataxia.
Entre otras causas menos comunes podríamos encontrar las anomalías congénitas, infecciones, otras enfermedades autoinmunes, Virus de la Inmunodeficiencia Humana, enfermedad de Creutzfeldt-Jakob, etc. En líneas generales, para que se produzca ataxia, estas patologías deben ocasionar daños en el cerebelo o a las estructuras relacionadas con éste como la médula espinal, tálamo o ganglios de la raíz dorsal. Una causa muy frecuente de ataxia es la hemorragia cerebelosa.
La anamnesis, el estudio del caso y la selección adecuada de pruebas diagnósticas son necesarios para encontrar la etiología correcta. El tratamiento irá enfocado a la intervención de la patología adquirida y el pronóstico dependerá de la gravedad de las lesiones.
2. Ataxias hereditarias recesivas
A diferencia de las ataxias adquiridas, estos tipos de ataxia suelen ser de inicio precoz, durante la infancia o entre los 20 y 30 años. Que la enfermedad sea recesiva implica que debemos haber heredado dos copias iguales del gen “defectuoso” de nuestros padres.
Esto implica que mucha población es simplemente portadora de la enfermedad aunque no se manifieste, ya que con un gen “sano” es suficiente para no desarrollarla. En este grupo encontramos algunos de los tipos más habituales de ataxia como la Ataxia de Friederich o la Ataxia-Telangiectasia.
2.1. Ataxia de Friederich
Es el tipo de ataxia hereditaria más común. Se estima que su prevalencia en los países desarrollados es de 1 persona por cada 50.000 casos. Su inicio suele ser en la infancia, presentando problemas en la marcha, torpeza, neuropatía sensorial y anormalidades en el movimiento de los ojos. Otras consecuencias menos frecuentes pueden ser deformaciones en el esqueleto y miocardipatía hipertrófica.
A medida que la enfermedad progresa, la disartria –alteración en la articulación de las palabras– , disfagia –dificultad en la deglución–, la debilidad en las extremidades inferiores, etc. son más aparentes. Se estima que entre los 9 y 15 años del inicio de los síntomas la persona pierde la capacidad de andar.
Este cuadro clínico es consecuencia de la neurodegeneración de las células ganglionares de la raíz dorsal, los tractos espinocerebelosos, las células del núcleo dentado –un núcleo profundo del cerebelo– y los tractos córticoespinales. Las células de Purkinge –las principales células del cerebelo– no resultan afectadas. El estudio de neuroimagen no suele mostrar ninguna afectación aparente del cerebelo.
Actualmente no existe cura y los tratamientos administrados suelen ser sintomáticos. El riesgo debido a la disfagia, la cardiomiopatía, etc., conlleva que se deba hacer un seguimiento habitual de los pacientes. Diversos ensayos clínicos están en marcha para observar el potencial de diversos fármacos como, entre otros, el interferon-gamma.
2.2. Ataxia-Telangiectasia
Con una prevalencia estimada de 1 caso entre 20.000-100.000 casos, la ataxia-telanigectasia (AT) es la causa más común de ataxia recesiva en pacientes menores de 5 años. A medida que la enfermedad se desarrolla podemos encontrar hipotonia –disminución del tono muscular–, polineuropatía –afectación del sistema nervioso periférico–, apraxia oculomotora –problemas en cambiar la mirada hacia un estímulo que debe fijarse–, etc. Los pacientes con AT suelen presentar inmunodeficiencias que ocasionan infecciones pulmonares recurrentes.
En el estudio de neuroimagen se puede observar atrofia del cerebelo, a diferencia de la ataxia de Friederich. Como en el caso anterior el tratamiento va dirigido a los síntomas y no existe una cura.
2.3. Otras ataxias hereditarias recesivas
Encontramos muchos más tipos de ataxias hereditarias como la Ataxia con apraxia oculomotora, la Ataxia Cayman, Ataxia con deficiencia de vitamina E, Ataxia espinocerebral infantil, etc.
3. Ataxia hereditarias dominantes
Las ataxias hereditarias dominantes ocurren en cada generación de una familia con un riesgo del 50% de recibir la enfermedad por parte de uno de los padres. En este caso, con una sola copia del gen afectado es suficiente para desarrollar la enfermedad. En función del curso de la enfermedad pueden ser divididas en episódicas o progresivas. Existen distintos test genéticos para el diagnóstico de estas patologías. Como en los casos anteriores, tampoco existen curas.
Ataxia y Apraxia: no son lo mismo
Desde un punto de vista neuropsicológico, el mayor diagnóstico diferencial que se debe realizar es distinguir la ataxia de la apraxia. Aunque puedan conllevar déficits cognitivos parecidos, sobre todo en las formas adquiridas, son significativamente diferentes desde un punto de vista clínico. La apraxia se define como una alteración en la ejecución de determinados movimientos aprendidos en respuesta a una orden y fuera del contexto que no es atribuible a afectaciones sensoriales o motoras, falta de coordinación o déficits atencionales.
La ataxia, en cambio, es un déficit motor de coordinación como tal. Aunque un paciente no pueda ejecutar la acción requerida ante una orden, será debido a la incapacidad motora. En la apraxia el problema surge porque el “input verbal” –es decir, la orden– no se puede asociar con la respuesta motora o “output motor”.
Por otro lado**, en la apraxia no deberíamos encontrar otros problemas como la inestabilidad de la marcha**, problemas en la deglución, etc. Así pues, en estos casos la evaluación neurológica será obligatoria si observamos signos incompatibles con la apraxia. No obstante, también cabe tener en cuenta que ambas manifestaciones clínicas pueden presentarse de manera concomitante.
La incidencia de la ataxia a nivel nacional
Con las prevalencias que hemos citado en el caso de la ataxia en su forma hereditaria, podemos considerar estas enfermedades como raras –siendo en Europa una enfermedad rara aquella que se da un caso cada 2000 personas–. Cuando las enfermedades se califican como raras generalmente es más difícil avanzar en su investigación para encontrar tratamientos eficaces.
Además, como hemos visto, las formas hereditarias de la enfermedad afectaran mayoritariamente a población infantil y joven. Esto ha hecho que surjan diversas asociaciones sin ánimo de lucro que promuevan el tratamiento, la divulgación y la mejora de calidad de vida de estos pacientes. Entre ellas encontramos la Asociación Catalana de Ataxias Hereditarias, la Asociación Sevillana de Ataxias y la Asociación Madrileña de Ataxias.
Conclusiones
La ataxia, aunque poco prevalente en su manifestación hereditaria, es un trastorno que afecta a las actividades de la vida diaria y la independencia en la vida de muchas personas, especialmente en población joven. Además, las prioridades farmacéuticas y empresariales hacen que la investigación en este campo avance con lentitud, por lo cual las propuestas en cuanto a tratamiento se centran en los cuidados paliativos.
Es por eso que se debe divulgar su existencia y dar conocer sus afectaciones. Cada paso, por pequeño que sea, puede representar mejoras en la calidad de vida de estos pacientes, con el alivio para el sistema sanitario que esto supone. El estudio y desarrollo en detección precoz y la automatización de sistemas de tratamiento resultarán beneficiosos tanto para pacientes, familiares, cuidadores y profesionales sanitarios. Cuando avanzamos en estos campos, todos salimos ganando y, por esta razón, debemos dar a conocer y apoyar estas causas sociales.