
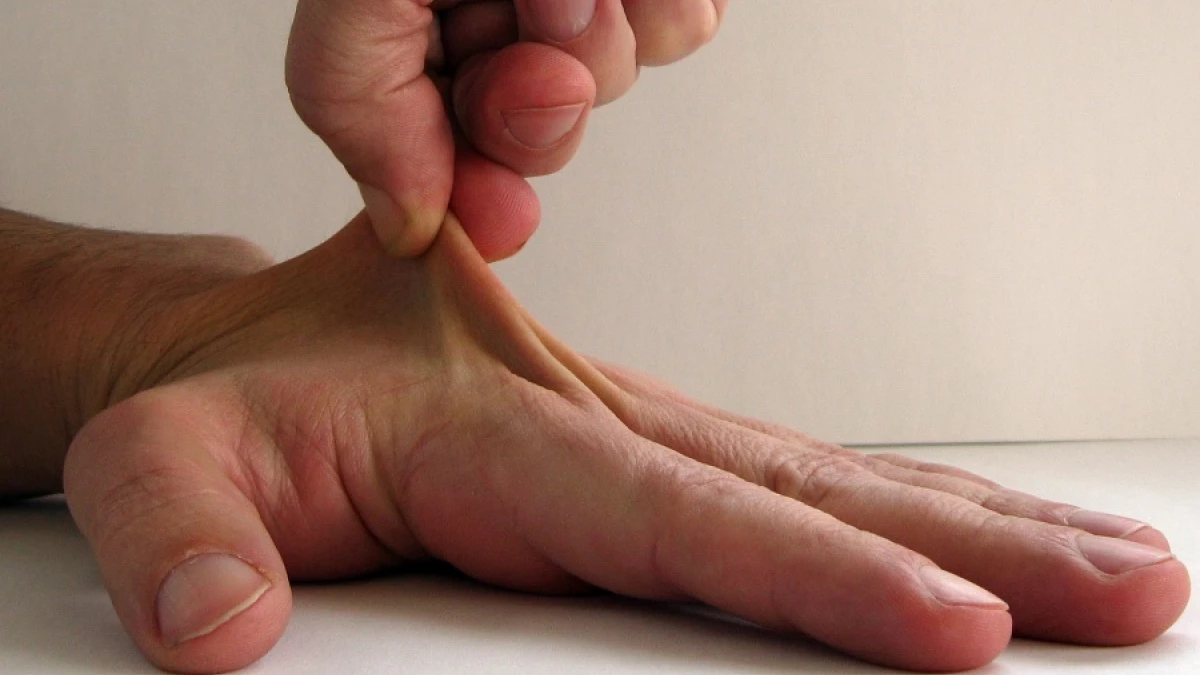

Posiblemente en alguna ocasión hayamos visto como alguien podía estirar su piel hasta límites poco habituales, o bien como algunas personas son capaces de hacer torsiones con diferentes partes de su cuerpo que escapan a la mayoría de las personas debido a que poseen hiperlaxitud.
En la mayoría de estos casos lo vemos como una curiosidad, y lo cierto es que alrededor de de un 10% de la población presenta esta característica sin tener mayor problema.
Sin embargo, existe un problema mucho más severo que en ocasiones comparte con las personas hiperlaxas algunas características, salvo que en su caso aparece junto a otros síntomas que suponen un perjuicio para su calidad de vida y que incluso pueden ser peligrosos para su supervivencia. Se trata del síndrome de Ehlers-Danlos, una extraña y poco habitual enfermedad genética de la cual vamos a hablar a lo largo de este artículo.
- Artículo relacionado: "Las diferencias entre síndrome, trastorno y enfermedad"
¿Qué es el síndrome de Ehlers-Danlos?
Recibe el nombre de síndrome de Ehlers-Danlos a un extraño y poco habitual síndrome, de origen genético, que se caracteriza por la presencia de alteraciones del tejido conjuntivo y concretamente por déficits en la producción de colágeno. Estos déficits, que se producen a nivel generalizada en todo el organismo, se traducen en una afectación a nivel de piel, articulaciones, vasos sanguíneos o incluso órganos. Se trata de una enfermedad principalmente observada en el ser humano, aunque también se han visto algunos casos en otros animales.
Aunque existe una gran heterogeneidad, entre los principales y más notorios síntomas de este trastorno podemos encontrar la presencia de hiperlaxitud en las articulaciones y ligamentos, hiperelasticidad de la piel y equimosis (lesiones en los que aparecen hemorragias dentro de la piel, como las que se producen con un golpe), así como la debilidad muscular y la fragilidad en los tejidos: no es infrecuente que aparezcan moratones al mínimo golpe, dolor en las articulaciones o facilidad para que aparezcan dislocaciones y luxaciones.
En algunos casos puede vincularse a artritis, problemas respiratorios y de visión. En casos graves, puede provocar rotura de órganos internos, problemas cardíacos o deformaciones, así como dolor crónico o tumores moluscoides.
El síndrome de Ehlers-Danlos es una enfermedad muy poco habitual, que solo padecen 1 de cada 5000 personas y que parece darse en mayor proporción en mujeres y niños. Es frecuente que este problema surja de manera comórbida con otros problemas como fatiga crónica, tendinitis, hipoacusia, fibromialgia o escoliosis. En algunos casos es confundida con celiaquía o incluso con abusos. A medida que los sujetos crecen, la hiperlaxitud tiende a bajar, pero el dolor y las complicaciones derivadas de ésta permanecen.
Se trata de un síndrome cuya variedad puede variar, pasando desde un problema leve hasta, y especialmente en algunos subtipos, poder tener repercusiones mortales. Esto es especialmente relevante en los casos con problemas vasculares o en los órganos, en los que puede haber roturas de los vasos sanguíneos o las paredes de intestinos o el útero (en este caso puede ser especialmente delicado el embarazo y el parto).
Aunque en la mayoría de casos la esperanza de vida es la normal y no existe disminución, los síntomas y sus posibles complicaciones pueden perjudicar y disminuir la calidad de vida.
- Quizás te interese: "Tipos de células principales del cuerpo humano"
Principales tipos
El síndrome de Ehlers-Danlos no es un trastorno homogéneo, sino que presenta una alta heterogeneidad. De hecho, más que de un solo trastorno podríamos hablar de un conjunto de ellos, existiendo diversas tipologías.
Aunque antiguamente se consideraban la existencia de hasta trece variantes, posteriormente los tipos de síndrome fueron reclasificados y se redujeron a un total de 6 (perdiéndose o integrándose en otros algunos como el de la córnea frágil, el displásico espondiloqueiral, el musculocontractual, el tipo periodontitis o el generado por déficit de tenascina-X), que son los que se presentan a continuación.
1. Tipo clásico
El denominado tipo clásico es de entre todas las variantes del síndrome la más común, y se caracteriza por la hipermovilidad de las articulaciones y la hiperelasticidad y extensibilidad de la piel, junto a la fragilidad de estos tejidos. Es habitual que puedan doblarse completamente los dedos y que se presenten dislocaciones, luxaciones y esguinces, y pueden darse neoplasias benignas.
Pequeñas lesiones suelen generar grandes hematomas, y acontecimientos como el embarazo pueden ser peligrosos. Pueden aparecer insuficiencias mitrales como consecuencia de deformaciones en las válvulas cardíacas, además de ser frecuentes las hernias en distintos puntos del tubo digestivo.
2. Tipo hipermóvil
Se trata del segundo tipo más habitual, y en este caso observamos hiperelasticidad y movilidad de articulaciones (las cuales se dislocan con facilidad) especialmente en áreas como brazos y piernas, así como con frecuencia dolor e hipersensibilidad a este. Al contrario que en el anterior tipo, no es tan habitual la fragilidad de la piel o los tejidos.
3. Tipo vascular
Probablemente el más grave y peligroso de todos los subtipos, aunque afortunadamente menos frecuene que los anterioes, es el síndrome de Ehlers-Danlos tipo vascular.
En este tipo la piel no es elástica y no existe hiperlaxitud en las articulaciones (salvo quizás en los dedos), pero tanto esta como otros tejidos son finos y frágiles (no es infrecuente poder ver las venas a través de la piel). Especialmente y tal como se puede adivinar por el nombre destaca la fragilidad de las arterias y otros vasos sanguíneos así como de los órganos, la cual provoca una gran facilidad para su laceraciones y ruptura.
Este es el más letal de todos los subtipos y el único que reduce la esperanza de vida, siendo la causa de la muerte por lo general que se rompan las venas y arterias de intestinos o útero (también en este caso el riesgo durante el embarazo es elevado).
4. Tipo cifoescoliótico
Un subtipo muy poco común (en el que apenas existen casos diagnosticados) el cual presenta características semejantes al clásico, con la característica añadida de la presencia de una escoliosis congénita que va a peor con el paso del tiempo.
Es habitual que exista una cada vez mayor debilidad a nivel muscular, osteopenia, y es posible que aparezca un retraso psicomotor. En algunos casos pueden llegar a acabar perdiendo la capacidad de caminar.
Puede conllevar síntomas típicos del síndrome de Marfan, desde la morfología típica (extremidades extremadamente largas) incluyendo el riesgo de afectación de la arteria aorta. También pueden tener problemas visuales, incluyendo la rotura del globo ocular, aunque esto no es tan habitual.
5. Tipo artrocalásico
Otro de los subtipos poco habituales, se caracteriza por presentar hiperlaxitud en las articulaciones, osteopenia y, por norma general, presencia de dislocaciones congénitas o frecuentes en ambas caderas. También tiende a haber hipotonía.
6. Tipo dermatosparáxico
Posiblemente el menos común y con apenas casos reconocidos, este subtipo se caracteriza por la fragilidad y laxitud cutánea, con herniaciones frecuentes y con pérdida de elasticidad. Es muy habitual la facilidad para sufrir hematomas.
¿Cuáles son sus causas?
El síndrome de Ehlers-Danlos es como hemos dicho un trastorno, o mejor dicho conjunto de trastornos, de origen genético. Así, los problemas en la síntesis de colágeno que generan la mayoría de los síntomas antes citados se derivan de la presencia de mutaciones en diferentes genes del organismo.
Algunos de los genes cuyas mutaciones se han asociado a este tipo de síndrome son COL5A1, COL5A2, ADAMTS2, COL1A1, COL1A2, PLOD2 o TNXB. Pese a ello, en algunos casos se desconoce qué alteraciones genéticas pueden estar produciendo el problema.
Aunque existen casos de novo en personas sin antecedentes familiares, los hallazgos parecen indicar que en muchos casos estamos ante un trastorno heredado. En los subtipos más habituales la herencia es de tipo autosómico dominante (especialmente en el clásico o el hipermóvil), mientras que en los menos frecuentes la herencia es autosómica recesiva (algo que ocurre en el tipo dermatosparaxis o en los tipos vascular o de cifoescoliosis).
Tratamiento de este síndrome
El síndrome de Ehlers-Danlos es un trastorno de origen genético que no cuenta con un tratamiento curativo, siendo los tratamientos existentes aquellos dirigidos a corregir o paliar los síntomas y alteraciones que genera.
Cada caso concreto requerirá de un tratamiento concreto y especializado. Por ejemplo, se puede realizar terapia ocupacional o fisioterapia para mejorar dificultades motoras o retrasos psicomotores, o bien emplear cirugía para corregir problemas cardíacos, drenar hemorragias, instalar prótesis o sujeciones o retirar tumoraciones.
Además de ello hay que tener en cuenta que tanto los pacientes como su entorno pueden presentar problemas psicológicos tales como ansiedad, depresión, problemas de autoestima y dificultades en diferentes ámbitos de su día a día. En este sentido puede ser de utilidad la psicoeducación y la aplicación de diferentes terapias psicológicas según el caso.













